Anticuerpos
monoclonales en enfermedades alérgicas: desarrollo, farmacología y
aplicaciones clínicas
Monoclonal antibodies in
allergic diseases: development, pharmacology, and clinical applications
Anticorpos
monoclonais em doenças alérgicas: desenvolvimento, farmacologia e aplicações
clínicas
Selene Pury1,2, Ricardo José Saranz1,
María José Irastorza1, Laura Veronica Sasia1, Pilar
Visconti1, Graciela Alegre1, Natalia Andrea Lozano1,
Yanina Viviana Berardi1, Alejandro Lozano1
DATOS DE AUTORES
1.
Universidad Católica de Córdoba, Facultad de Ciencias de la Salud,
Cátedra de Inmunología; Clínica Universitaria Reina Fabiola, Servicio de
Alergia e Inmunología, Córdoba, Argentina.
2.
Mail
de contacto: selenepury@gmail.com.
CONCEPTOS CLAVE
¿Qué
se sabe sobre el tema?
Las
enfermedades alérgicas se manifiestan con múltiples endo-fenotipos. La
introducción de los fármacos biológicos en la última década ha contribuido a
su abordaje terapéutico desde la medicina de precisión. Su desarrollo está en
continuo avance.
¿Qué
aporta este trabajo?
Este artículo
aporta los conceptos históricos, bases farmacológicas y las evidencias más
actuales sobre el uso de medicamentos biológicos en patologías como asma, urticaria
crónica idiopática, rinosinusitis crónica con poliposis nasal y dermatitis atópica
graves.
Divulgación
Las enfermedades alérgicas
constituyen un grupo heterogéneo de patologías de alta prevalencia y gran
impacto en la calidad de vida, particularmente en sus formas graves. Los
avances en el conocimiento de los mecanismos fisiopatogénicos celulares y
moleculares nos han permitido el desarrollo de tratamientos con fármacos biológicos
muy evolucionados que, indicados correctamente y mantenidos en el tiempo,
contribuyen a mejorar sensiblemente la calidad de vida de los pacientes con
alergias graves.
Anticuerpos monoclonales en
enfermedades alérgicas: desarrollo, farmacología y aplicaciones clínicas
Resumen
El avance en la comprensión de
los procesos inmunológicos relacionados con enfermedades alérgicas y en la
bioingeniería de anticuerpos ha propiciado el desarrollo de terapias biológicas
específicas. Los anticuerpos monoclonales, dirigidos selectivamente hacia
citocinas involucradas en la patogénesis de los procesos alérgicos, o sus
receptores, han emergido como una herramienta prometedora en el tratamiento de
diversas afecciones, tales como el asma, la rinitis alérgica, la urticaria y la
dermatitis atópica grave. Desde la aprobación del primer anticuerpo monoclonal
anti-CD3 de origen murino en 1986, se ha transitado un largo recorrido,
caracterizado por la aparición de anticuerpos quiméricos, los que por estructura
molecular de la inmunoglobulina es semejante a los del ser humano denominados “humanizados”
y los exactamente iguales llamados “completamente humanos”. La “humanización”
de los anticuerpos monoclonales ha contribuido significativamente a reducir
tanto el riesgo de inmunogenicidad como la incidencia de efectos adversos,
mejorando notablemente la seguridad y eficacia de estas terapias.
Este artículo tiene como
objetivo abordar la caracterización, desarrollo, farmacocinética,
farmacodinamia y utilidad clínica de los anticuerpos monoclonales,
principalmente focalizados en las enfermedades alérgicas.
Palabras clave: inmunoglobulinas;
alergia; asma; rinitis alérgica; farmacología
Monoclonal antibodies in allergic diseases:
development, pharmacology, and clinical applications
Abstract
The understanding of immunological processes associated
with allergic diseases and advancements in antibody bioengineering has driven
the development of specific biological therapies. Monoclonal antibodies,
selectively targeting cytokines involved in the pathogenesis of allergic
processes or their receptors, have emerged as a promising tool in treating
various conditions, including asthma, allergic rhinitis, urticaria, and severe
atopic dermatitis. Since the approval of the first anti-CD3 mouse monoclonal
antibody in 1986, remarkable progress has been achieved, marked by the
development of chimeric, 'humanized,' and 'fully human' antibodies. The
'humanization' of monoclonal antibodies has played a crucial role in reducing
the risk of immunogenicity and minimizing adverse effects, thereby notably
enhancing the safety and efficacy of these therapeutic interventions.
The aim of this article is to address the
characterization, development, pharmacokinetics, pharmacodynamics, and clinical
utility of monoclonal antibodies, with a primary focus on allergic diseases.
Keywords: immunoglobulins;
hypersensitivity; asthma; allergic rhinitis; pharmacology
Anticorpos monoclonais em doenças
alérgicas: desenvolvimento, farmacologia e aplicações clínicas
Resumo
O avanço na compreensão dos
processos imunológicos relacionados a doenças alérgicas e na bioengenharia de
anticorpos tem impulsionado o desenvolvimento de terapias biológicas específicas.
Os anticorpos monoclonais, direcionados seletivamente para citocinas envolvidas
na patogênese de processos alérgicos ou seus receptores, emergiram como uma
ferramenta promissora no tratamento de diversas condições, incluindo asma,
rinite alérgica, urticária e dermatite atópica grave. Desde a aprovação do
primeiro anticorpo monoclonal murino anti-CD3 em 1986, notáveis avanços foram
alcançados, marcados pelo desenvolvimento de anticorpos quiméricos,
"humanizados" e "totalmente humanos". A "humanização"
dos anticorpos monoclonais desempenhou um papel crucial na redução do risco de
imunogenicidade e na minimização de efeitos adversos, aumentando
significativamente a segurança e eficácia dessas intervenções terapêuticas.
O objetivo deste artigo é abordar
a caracterização, desenvolvimento, farmacocinética, farmacodinâmica e utilidade
clínica de anticorpos monoclonais, com foco principal em doenças alérgicas.
Palavras-chave: imunoglobulinas;
hipersensibilidade; asma; rinite alérgica; farmacologia
Introducción
Los anticuerpos monoclonales (mAb), que se dirigen a
citocinas involucradas en la patogénesis de las enfermedades alérgicas, han
emergido como una herramienta prometedora en el tratamiento de diversas
patologías como el asma, rinitis alérgica, urticaria y dermatitis atópica
graves.
El uso de anticuerpos para el tratamiento de enfermedades se
remonta a la década de 1890, cuando Emil von Behring descubrió que pequeñas
dosis de toxina diftérica o tetánica tenían el potencial de generar una
inmunidad que podía transferirse de un animal a otro a través del suero.1 Recién a principios de la
década de 1960 se describieron las características estructurales de los
anticuerpos. Fue el trabajo de Rodney R. Porter, quien, tras combinar sus
descubrimientos con los hallazgos de Gerald M. Edelman, culminó con la
publicación del primer modelo de la estructura de la inmunoglobulina G.2 En
1975, el investigador argentino César Milstein y Georges Köhler, en la
Universidad de Cambridge, fusionaron por primera vez linfocitos B esplénicos de
animales hiperinmunizados con células murinas de un mieloma y obtuvieron un
hibridoma: una línea celular monoclonal, inmortal capaz de producir anticuerpos
específicos siempre para el mismo epítope antigénico.3
Desde su primera administración en forma de sueros, se ha
recorrido un largo camino hasta la actualidad, con el desarrollo de anticuerpos
monoclonales, fragmentos de anticuerpos, anticuerpos de dominio y anticuerpos
policlonales.
El objetivo de esta revisión es abordar la caracterización,
desarrollo, farmacocinética, farmacodinamia y utilidad clínica de los
anticuerpos monoclonales, dirigidos a las enfermedades alérgicas.
Materiales y métodos
Se realizó una búsqueda bibliográfica que abarcó
tanto fuentes en inglés como en español, utilizando las bases de datos
electrónicas de Medline y Latindex. Para optimizar la búsqueda, se emplearon
los términos MeSH “monoclonal antibody”, “immunoglobulins”, “biologic drugs”
“hypersensitivity”, “asthma”, “rhinitis”, “chronic urticaria”, “pharmacology” y
sus correspondientes DeCS en español. La búsqueda incluyó artículos
de revisión, observacionales,
analíticos y guías de práctica clínica. Se incluyeron estudios
publicados entre 2013 y 2023. Para el apartado de historia y la generación de
los anticuerpos monoclonales, se realizó una
revisión narrativa de artículos
con fechas previas, en especial los estudios que representaron los primeros
hallazgos sobre el tema. Se
excluyeron artículos en idioma
diferente al inglés y español, publicaciones por duplicación de información o a
la que no se pudo acceder a texto completo. La búsqueda obtuvo un total de 310
estudios potenciales que, posteriormente a la
eliminación, considerando
los criterios de
aceptación y exclusión,
permitió la selección de 50 artículos incluidos en el apartado de referencias.
Desarrollo de los
anticuerpos monoclonales
Los mAb y sus proteínas derivadas se han
consolidado como uno de los grupos más grandes de biológicos o proteínas
bioterapéuticas.4 La tecnología del hibridoma de ratón descrita por
Milstein y Köhler fue un paso importante en el desarrollo de la tecnología de
anticuerpos y allanó el camino para la aparición de anticuerpos monoclonales
terapéuticos. (Fig. 1).
En 1986,
se aprobó el primer mAb, muromonab-CD3, de origen murino del tipo IgG2a que fue
creado mediante la tecnología del empleo de hibridomas.4,5 Este
anticuerpo específico reconoce la molécula del receptor CD3 presente en la
superficie de los linfocitos T, y su mecanismo de acción consiste en bloquear
la unión del receptor de células T (TCR) a los antígenos, lo que lo convierte
en una terapia inmunosupresora. Se aprobó para el tratamiento del rechazo agudo
del trasplante renal resistente a los corticoides.5
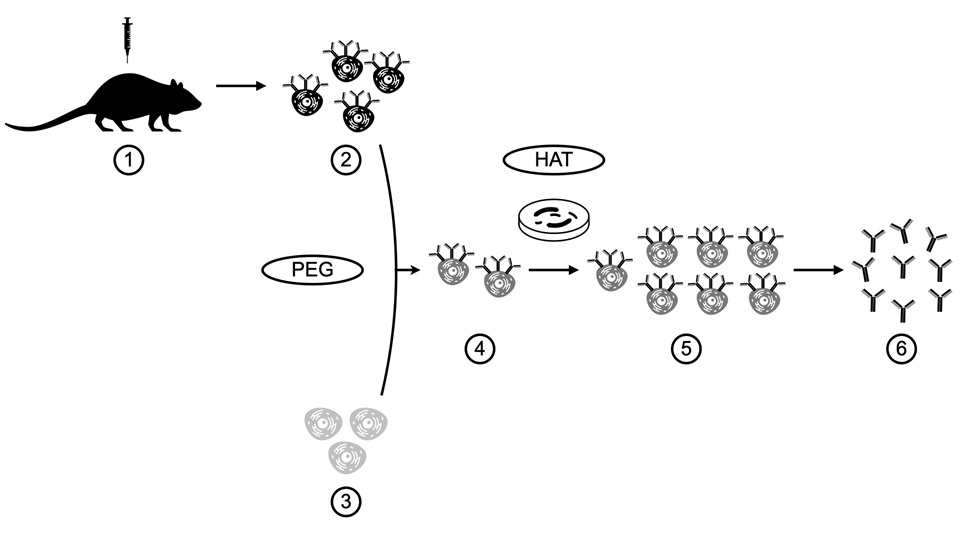
Figura 1:
Proceso de formación de hibridomas: 1) Se inmuniza a un animal, generalmente un
ratón o una rata, con el antígeno de interés. 2) Extracción de células del bazo
del animal inmunizado. 3) Células de mieloma múltiple o plasmocitoma de rata o
ratón 4) Formación del hibridoma: Las células B de los animales inmunizados se
fusionan con las células tumorales, que tienen la capacidad de crecimiento
indefinido en cultivo y producir anticuerpos, esta fusión es favorecida por el
uso de Polietilenglicol (PEG) 5) Después de la fusión, las células se cultivan
en HAT (medio que contiene hipoxantina-aminopterina-timidina) que solo permite
sobrevivir a los hibridomas. Se seleccionan las células híbridas que se han
formado con éxito y se clonan para obtener poblaciones de células idénticas que
producen el mismo anticuerpo monoclonal 6) Anticuerpos monoclonales. Los
anticuerpos monoclonales se purifican
No obstante, los mAb de origen murino demostraron
tener limitaciones en su aplicación como agentes terapéuticos. Esto se debió,
en parte, a su corta vida media en la circulación sanguínea y a la inducción de
anticuerpos humanos contra anticuerpos murinos. La respuesta inmune contra los
anticuerpos de muromonab-CD3 inhibe su unión a CD3 y puede llevar al fracaso
terapéutico.6 A partir de la experiencia clínica adquirida con el
mAb anti-CD3 murino, se evidenció que la respuesta inmunológica del huésped,
junto con la ineficiente
farmacocinética de los anticuerpos murinos en
seres humanos, constituían obstáculos significativos para lograr una eficacia
sostenida en tratamientos a largo plazo.7 Además, se han descrito
reacciones vasculíticas mediadas por inmunocomplejos y anafilácticas por la
presencia de IgE circulante, en pacientes sensibilizados.8 Esto
impulsó el desarrollo de la producción de mAb quiméricos, es decir,
monoclonales con dominios constantes humanos y variables de roedores. El primer
mAb quimérico aprobado fue el Abciximab (inhibidor de agregación plaquetaria)
en 1994, seguidos de Rituximab (anti-CD20), Basiliximab (anti-CD25), Cetuximab
(Anti-EGFR), Infliximab (Anti TNFα), entre otros.4 La “humanización”
de los anticuerpos monoclonales ha disminuido, pero no eliminando por completo,
el riesgo de inmunogenicidad en comparación con los anticuerpos de ratón.9
La siguiente fase de desarrollo se diseñó para
generar mAbs “humanizados” mediante la sustitución de secuencias genéticas de
roedores por secuencias humanas. (Fig. 2) El primer anticuerpo monoclonal
“humanizado” aprobado fue el daclizumab, en 1997, dirigido contra la subunidad
Tac del CD25, receptor de la interleucina 2. Adalimumab fue el primer
anticuerpo monoclonal “completamente humano” anti-factor de necrosis tumoral
alfa aprobado para el tratamiento de la artritis reumatoidea.4,10 La
generación de anticuerpos monoclonales terapéuticos “completamente humanos” se
hizo posible gracias al desarrollo de plataformas de presentación de fagos o
“Phage display” y, más recientemente, mediante plataformas de ratones
transgénicos.
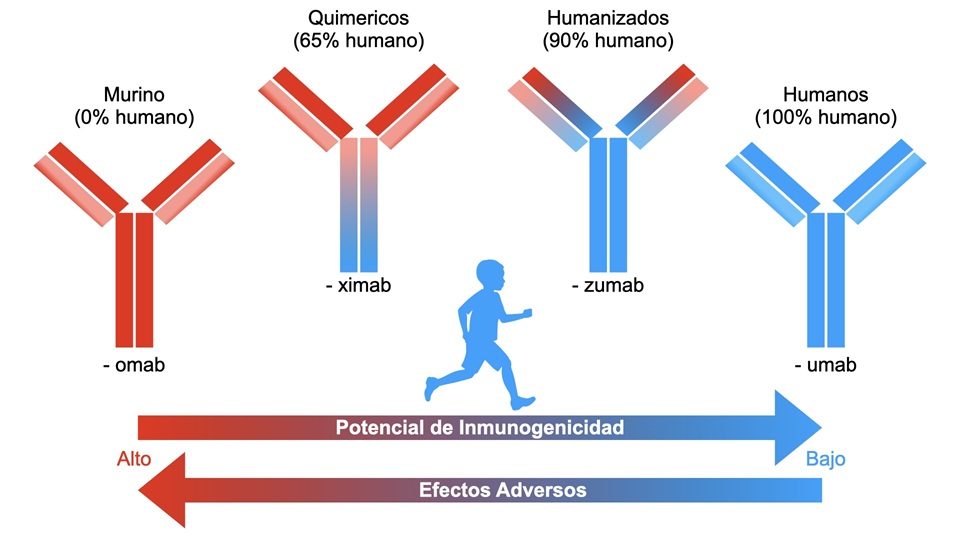
Figura 2:
Descripción esquemática de la “humanización” de anticuerpos desde anticuerpos
murinos (dominios rojos) hasta anticuerpos completamente humanos (dominios azules)
y sus sufijos asociados.
La tecnología de “Phage display” se basa en la
capacidad de exponer un ligando (péptido o proteína) en la cápside de un
bacteriófago. En 1985, Smith demostró que manipulando el genoma de los fagos
con la técnica de ADN recombinante, mediante la fusión del gen del ligando al
gen que codifica una proteína de la cápside, se podían obtener partículas con
péptidos fusionados a proteínas de su cápside.11 Esta técnica utiliza
bacteriófagos filamentosos pertenecientes a la familia del género Inovirus,
como el Fd o M13. Estos
bacteriófagos infectan cepas de Escherichia coli al unirse al receptor
pilus F de la bacteria, mediado por la proteína pIII, y posteriormente se translocan
hacia el citoplasma de la célula. Las proteínas de fago más comúnmente
utilizadas son la proteína pIII y pVIII. Estos fagos, modificados por
ingeniería genética, incorporando a su material genético genes foráneos de
interés, logran expresar en su superficie diferentes péptidos o moléculas que
antagonizan funciones específicas del sistema inmune con interés terapéutico.
El objetivo de esta tecnología es lograr una molécula con afinidad máxima por
otra molécula blanco. Los anticuerpos no son expresados en el fago en forma de
una inmunoglobulina entera sino en forma de fragmentos Fab o fragmentos
variables monocatenarios. (Fig 3).12-14
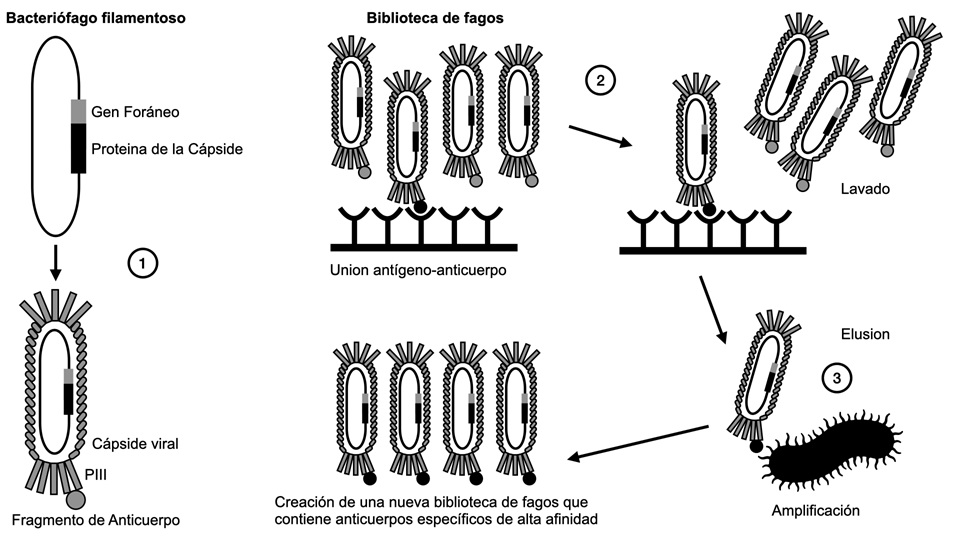
Figura
3: Plataformas de
presentación de fagos ("phage display") 1- Inicialmente, se aíslan
linfocitos B de donantes humanos, y se extraen y amplifican los genes que
codifican las cadenas variables de los anticuerpos de interés, compuestas por
cadenas pesadas (VH) y livianas (VL). Estas secuencias genéticas, tras
recombinación y randomización, generan todas las combinaciones posibles de VH y
VL, siendo luego insertadas en el genoma del bacteriófago, formando así una
biblioteca de fagos. 2- "Panning": Los fagos se ponen en contacto con
el antígeno de interés inmovilizado en una superficie sólida. Aquellos que se
unen con alta afinidad al antígeno se retienen, mientras que los que no se unen
son lavados. 3- Los fagos que están unidos al antígeno se eluyen y se utilizan
para infectar Escherichia coli, amplificando así la cantidad de fagos que exhiben
fragmentos de anticuerpos específicos. Estos fragmentos se incorporan al
esqueleto estructural del anticuerpo, formando anticuerpos completos.
Por otro lado, el uso de animales genéticamente
modificados ha permitido desarrollar nuevos anticuerpos monoclonales
“completamente humanos”. Esta tecnología se introdujo en 1994 con el desarrollo
de dos líneas de ratones transgénicos “HumAb” mouse15 y “XenoMouse”.16 Se emplean
ratones genéticamente modificados mediante técnicas de manipulación genética
dirigida, permitiendo que estos animales sean capaces de sintetizar Igs
completamente humanas, con altas afinidades y especificidades de unión al
antígeno objetivo.17-18 Panitumumab, anticuerpo monoclonal inhibidor
del receptor del factor de crecimiento epidérmico fue el primer anticuerpo
desarrollado con esta tecnología, aprobado por la FDA de Estados Unidos en 2006
(Fig. 4).10,19
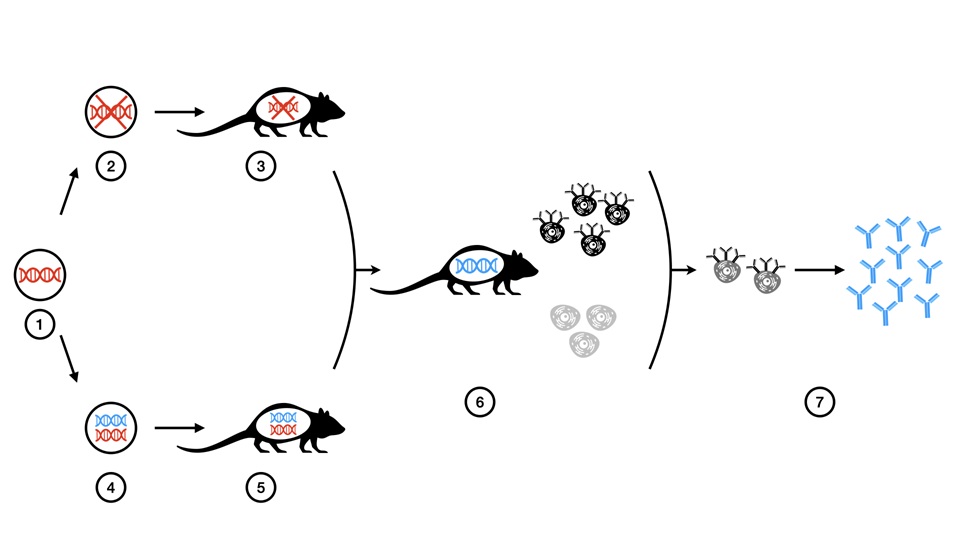
Figura 4:
Generación de anticuerpos monoclonales a partir de ratones transgénicos
"XenoMouse". 1) célula madre embrionaria de ratón 2) Los loci de los
genes de inmunoglobulinas de ratón (cadenas pesadas y ligeras) fueron
inactivados en células madre embrionarias mediante la eliminación dirigida de
genes 3) Se genera una cepa de ratón incapaz de producir anticuerpos 4) Célula
madre embrionaria de ratón en la cual se introdujeron genes de anticuerpos
humanos 5) Se logra una cepa de ratón transgénico capaz de producir tanto
anticuerpos humanos como de ratón. 6) Los ratones incapaces de producir
inmunoglobulina de ratón se cruzan con los ratones transgénicos, lo que resulta
en la cepa de “XenoMouse” que expresa anticuerpos completamente humanos y es
incapaz de producir anticuerpos de ratón. 7) Las células B productoras de
anticuerpos, aisladas del bazo de un “XenoMouse” inmunizado, se utilizan para
producir hibridomas. Cada hibridoma puede producir grandes cantidades de
anticuerpos completamente humanos
Actualmente se han desarrollado estrategias de
desarrollo de anticuerpos monoclonales humanos basados en la amplificación
directa de los genes que codifican las regiones VH y VL de células B humanas y
su posterior expresión en sistemas de cultivo celular. La ventaja de este
método es que permite el aislamiento y la formación de anticuerpos humanos
nativos con el emparejamiento natural de VH y VL, lo que no es posible en la
tecnología de visualización de fagos y animales transgénicos.20
Mecanismo de acción
Junto con el cáncer, las enfermedades
inflamatorias, entre ellas las enfermedades alérgicas
y autoinmunes son el enfoque de las terapias con
anticuerpos monoclonales. El mecanismo de acción varía según el objetivo
terapéutico. Sin embargo, sus funciones se pueden resumir en tres modos
principales de acción: bloqueando factores solubles, actuando sobre receptores
celulares específicos o actuando como moduladores de la respuesta inmune.21
La capacidad neutralizante de los anticuerpos
monoclonales permite disminuir la concentración efectiva de moléculas solubles
específicas. En el caso de enfermedades inflamatorias, las terapias basadas en
anticuerpos están diseñadas para unirse a citocinas específicas o para bloquear
su unión a receptores. Las citocinas asociadas con la inflamación y
autoinmunidad incluyen el factor de necrosis tumoral alfa (TNF-α), la mayoría
de las interleucinas y de las proteínas del complemento.22
Los anticuerpos monoclonales también pueden
actuar regulando el sistema inmunológico, por ejemplo, dirigiéndose a
receptores de la superficie celular, como CD20 y CD4 en células B o T
respectivamente. Además, los mAbs pueden ejercer su acción mediante
citotoxicidad celular dependiente de anticuerpos, donde las células
responsables del efecto son aquellas que presentan un receptor para la región
Fc del anticuerpo. Otro método indirecto por el cual los anticuerpos pueden
promover la muerte celular es la citotoxicidad dependiente del complemento, en
la cual la unión de los anticuerpos a la célula objetivo activa la cascada del
complemento.23
Absorción
En cuanto a la vía de administración, los mAb se
administran generalmente por vía intravenosa. Esta forma de administración
permite lograr una disponibilidad sistémica completa, un rápido ingreso de los
anticuerpos en la circulación y la obtención de concentraciones farmacológicas
elevadas. También es posible su administración por vía subcutánea o
intramuscular. No obstante, en la vía extravascular la absorción puede variar
según la cantidad de degradación pre-sistémica causada por enzimas
proteolíticas. La biodisponibilidad por vía subcutánea es de un 52-80%.24
La absorción sistémica de anticuerpos después de la administración subcutánea o
intramuscular ocurre a través del sistema linfático lentamente y las
concentraciones plasmáticas máximas de anticuerpos suelen observarse de 1 a 8
días después de la administración.24
La absorción oral de anticuerpos monoclonales
está limitada dado que pueden sufrir desnaturalización en el pH ácido del
estómago y degradación proteolítica en el estómago e intestino. Además, debido
a su gran tamaño molecular y alta polaridad, muestran tasas mínimas de difusión
a través del epitelio gastrointestinal. Sin embargo, las inmunoglobulinas
intactas pueden alcanzar la circulación sistémica al cruzar las células
epiteliales a través del transporte paracelular o mediante procesos mediados
por receptores. Por ejemplo, en niños recién nacidos, la IgG se absorbe
eficientemente después de la administración oral. Esta absorción
gastrointestinal permite la transmisión de IgG de la leche materna al recién
nacido y se da mediante el receptor de IgG, FcRn (receptor Fc del neonato).25
Se ha reportado que FcRn se expresa en una amplia variedad de tejidos en
adultos y protege la IgG de la degradación. Este receptor desempeña un rol
principal en la biodisponibilidad de los mAb. 24-26
Distribución
Debido a su elevado peso molecular y naturaleza
hidrofílica, la capacidad para difundir a través de las membranas biológicas es
escasa. El transporte por convección (ingreso de solutos a través de una
membrana impulsado por el paso del agua de un compartimento a otro) es el
mecanismo principal de extravasación, ya que más del 98% de los anticuerpos
ingresan a los tejidos a través de este proceso y ocurre como resultado del
movimiento de fluido desde la sangre hacia el espacio intersticial de los
tejidos, que es impulsado principalmente por el gradiente de presión
hidrostática entre la sangre y el tejido 26. Otros factores
determinantes del transporte por convección comprenden el gradiente de presión
osmótica y el diámetro de los poros paracelulares del endotelio vascular.26
El fluido que penetra en los tejidos mediante extravasación regresa al
torrente sanguíneo mediante el sistema de circulación linfática. Los vasos
linfáticos son mucho más grandes que los poros paracelulares del endotelio
vascular, lo que facilita un flujo convectivo de anticuerpos prácticamente sin
restricciones a través de la linfa. Debido a las diferencias en la eficiencia
de la captación convectiva en los tejidos y la eliminación de anticuerpos a
través del sistema linfático, las concentraciones de los mismos en el líquido
intersticial de los tejidos suelen ser inferiores a las concentraciones
plasmáticas.
La
velocidad de eliminación de los anticuerpos del tejido depende principalmente
de la depuración por eliminación convectiva y las tasas de catabolismo de los
anticuerpos en el propio tejido. Los anticuerpos muestran muy poca distribución
en el sistema nervioso central.27,28
Metabolismo y
excreción
La excreción renal de estos fármacos es limitada
debido a su gran tamaño molecular, lo que dificulta una filtración eficaz a
través del glomérulo. La principal vía de eliminación de los anticuerpos
monoclonales es a través del proceso de catabolismo intracelular, donde se
descomponen en aminoácidos después de ser captados por las células mediante
mecanismos de pinocitosis o endocitosis mediada por receptores.24 La
endocitosis mediada por receptor es un mecanismo dependiente de la interacción
de los dominios de unión Fab del anticuerpo con epítopes específicos presentes
en la superficie celular. Esta interacción conduce a la internalización
vesicular del anticuerpo para su posterior degradación. Este mecanismo se
conoce como "disposición mediada por la diana", y se caracteriza por
ser saturable, lo que implica un comportamiento y una cinética no lineal.29
Sin embargo, este mecanismo de disposición mediada por la diana no siempre
requiere que el dominio Fab del anticuerpo se una a un receptor en la
superficie celular. En algunos casos, sustancias solubles pueden unirse a dos o
más anticuerpos, formando grandes complejos que pueden ser eliminados mediante
fagocitosis.
Además, la endocitosis mediada por receptor
también puede ocurrir a través de los receptores Fcγ 25, y los
complejos formados por IgG y estos receptores (IgG-FcγR) tienen la capacidad de
desencadenar la endocitosis y el posterior catabolismo del anticuerpo.26
La pinocitosis es una endocitosis de fase fluida
inespecífica que no discrimina que proteínas son capturadas y sometidas a
degradación lisosomal. Sin embargo, la IgG se diferencia de la mayoría de las
proteínas debido a un mecanismo especial de reciclaje. Cuando la IgG ingresa al
entorno ácido de los endosomas celulares puede unirse a través de su fragmento
Fc al receptor FcRn, descrito por Brambell.30 La unión a FcRn
protege a la IgG de la degradación intracelular, permitiendo que se libere a la
sangre o al espacio intersticial. Aquella IgG que no se une a FcRn se somete a
la degradación proteolítica en el lisosoma. Se ha demostrado que la vida media
sérica de los anticuerpos IgG está directamente relacionada con su afinidad de
unión al FcRn.31
Además, la expresión de FcRn también se observa
en células epiteliales glomerulares renales humanas, lo que sugiere que FcRn
tenga un papel significativo en la reabsorción de IgG que ha sido filtrada,
contribuyendo así a reducir la importancia de la excreción urinaria como una
vía de eliminación primaria para la IgG. La afinidad de la IgG por FcRn es
específica de la especie. La baja afinidad del FcRn humano por la IgG de ratón
ayuda a explicar la rápida eliminación de mAbs murinos en humanos, con una vida
media de aproximadamente 1–2 días. Las vidas medias de los anticuerpos
monoclonales de IgG aumentan conforme se "humanizan". 24,26,28
Utilidad clínica en
enfermedades alérgicas
Dentro del grupo de las enfermedades
inflamatorias, las enfermedades de etiología alérgica son un blanco relevante
para el desarrollo de anticuerpos monoclonales. Los avances en la comprensión
de los mecanismos fisiopatológicos involucrados en las enfermedades alérgicas y
respiratorias han llevado a la posibilidad de una terapia más dirigida dentro
de la denominada "medicina de precisión". Esta, reconoce que, incluso
en pacientes con presentaciones clínicas similares de una enfermedad, los mecanismos
fisiopatológicos subyacentes pueden ser diversos, lo que genera diferentes
fenotipos y endotipos de una misma enfermedad.32
Asma
En la actualidad, los pacientes que presentan
asma alérgica moderada a grave pueden ser tratados utilizando diversos
anticuerpos monoclonales direccionados a citocinas inflamatorias claves
implicadas en la patogénesis de esta enfermedad. Sin embargo, el término asma
engloba diferentes endotipos. El eje principal de división se refiere al tipo
de inflamación, es decir, la inflamación de tipo 2 (T2 alto) y la inflamación
no tipo 2 (o T2 bajo).33 El mayor avance en terapia con anticuerpos
monoclonales ha emergido para el tratamiento del fenotipo T2 alto,
principalmente alérgico. Los datos clínicos existentes sobre anti-IgE,
anti-IL-5, anti- IL4-IL-13 así como anticuerpos dirigidos contra la
linfopoyetina estromal tímica (TSLP) y otras vías inmunológicas indican que
estas terapias generan tasas reducidas de exacerbación, una mejoría en la
función pulmonar, un mayor control del asma y una mejor calidad de vida.34,35
Así como disponemos en la actualidad de un gran
abanico de opciones terapéuticas con anticuerpos monoclonales para el
tratamiento del asma T2 alto o eosinofílico, para el tratamiento del asma con
un perfil T2 bajo las opciones con mAbs son más limitadas y los resultados no
son tan alentadores. En esta variante se describe con frecuencia un aumento de
los neutrófilos en el esputo y una activación de la vía de señalización de la
interleucina-17 (IL-17). Los agentes biológicos que se espera funcionen mejor
en pacientes con el fenotipo neutrofílico son aquellos que bloquean las
citocinas IL-1β, IL-6, IL-8, IL-17 e IL-23.36 No obstante, no se ha
establecido con certeza si la IL-17 es causal de la patología o si responde al
daño epitelial o a la colonización bacteriana de las vías respiratorias.37
El mayor ensayo clínico llevado a cabo con un
anticuerpo monoclonal anti-receptor de IL-17, el brodalumab, no mostró mejoría
en los síntomas ni en la función pulmonar 38 al igual que el
anticuerpo anti-IL-23, el risankizumab, que bloquea la diferenciación de las
células Th17.39 Existe
una necesidad no satisfecha de desarrollar nuevos tratamientos para pacientes
con asma neutrofílica, especialmente aquellos con enfermedad grave.
Urticaria crónica idiopática
La urticaria crónica idiopática, se define como
la urticaria presente durante al menos 6 semanas de origen no determinable. En
esta entidad, el infiltrado celular en su mayoría consiste en eosinófilos,
monocitos, basófilos y linfocitos T con un perfil Th2, que dependen de
moléculas como la TSLP, IL-33, IL-4, IL-13 y el receptor de prostaglandina D2
CRTH2 por lo que, se considera razonable el uso de anticuerpos monoclonales
dirigidos a estas moléculas.40
Omalizumab es el primer y único anticuerpo monoclonal humanizado IgG1
anti-IgE aprobado en 2014 por la Administración de Alimentos y Medicamentos
(FDA) para el tratamiento de la urticaria crónica idiopática. Este biológico se
une de forma específica a la inmunoglobulina IgE. La justificación de su uso se
basa en el papel clave de la IgE y su receptor de alta afinidad, FcεRI, en la degranulación
de los mastocitos de la piel.41
Existen ensayos prometedores con dupilumab42, mepolizumab43
y benralizumab.44
Dermatitis atópica
La dermatitis atópica es una enfermedad
inflamatoria crónica de la piel, cuyo principal síntoma es el prurito de
intensidad variable. La fisiopatología de la dermatitis atópica es compleja y
multifactorial, se entrelazan factores genéticos, defectos en la barrera
epidérmica, una respuesta inmunológica alterada y un desequilibrio en el
microbioma de la piel. Entre los factores inmunológicos involucrados se ha
evidenciado una activación de las células linfoides innatas tipo 2 (ILC2),
conocidas por su capacidad para secretar citoquinas proalérgicas moduladoras de
la respuesta inmune y proinflamatorias, como IL-4, IL-5, IL-9 e IL-13.45 A su vez, los factores implicados en
la inflamación de la epidermis, estimula a los queratinocitos a liberar
alarminas como TSLP, IL-25 e IL-33 promoviendo además la respuesta inmunológica
mediada por Th2.
La quimiotaxis de los linfocitos Th1 y el aumento
en la producción de las citoquinas IL-2, IL-12, TNFα e INF ocurrían durante la
fase crónica de la enfermedad con aumento de la respuesta de Th1 e
intensificación las respuestas de Th2 y Th22.46
La única terapia biológica aprobada en la
actualidad en Argentina para los pacientes con dermatitis atópica
moderada-grave es dupilumab que tiene como blanco la subunidad del receptor
alfa de la IL-4 que bloquea la señalización tanto de la IL-4 como de la IL-13.47 En fases de desarrollo se encuentran actualmente otros biológicos
dirigidos a IL-13, IL-31, IL-33, OX40, linfopoyetina estromal tímica e
inhibidores de la JAK quinasa 48
Rinosinusitis crónica alérgica y poliposis nasal
La patogénesis de la rinitis alérgica involucra
un proceso que abarca la sensibilización a alérgenos, la generación de IgE
específica, el reclutamiento de células inflamatorias y la desgranulación
mastocitaria. Se ha propuesto que los aeroalergenos, al entrar en contacto con
la mucosa nasal, inducen la generación de citocinas derivadas de las células
epiteliales, como el TSLP, IL-25 e IL-33 que median la activación de las
células linfoides innata, particularmente la ILC-2 con liberación de IL-4, IL-5
e IL-13. Los alérgenos también son captados por las células presentadoras de
antígenos que inducen la diferenciación de linfocitos T hacia un perfil TH2,
induciendo la producción de IgE.49
Otra patología nasal relacionada, pero distinta,
que implica un patrón de citocinas TH2 de inflamación es la rinosinusitis
crónica con poliposis nasal. Esta es una enfermedad heterogénea que requiere
abordajes tanto médicos como quirúrgicos en su tratamiento. La inflamación eosinofílica
y la liberación de mediadores tóxicos impulsados por un aumento en la expresión
de IL-5 y eotaxina desempeñan un papel crucial en el mecanismo del desarrollo
de los pólipos nasales. Están aprobados para su uso en pacientes con poliposis nasal crónica con rinosinusitis los
biológicos que se dirigen al receptor de IL-4 (dupilumab), a la IgE
(omalizumab) y a IL-5 (mepolizumab, reslizumab y benralizumab)50
En la tabla 1 se exponen los principales mAbs
aprobados y en fase III en Argentina para uso en enfermedades alérgicas.
|
TABLA 1. Anticuerpos
monoclonales disponibles en Argentina para su uso en enfermedades alérgicas.
|
|
Anticuerpo
|
Nombre comercial /Laboratorio
|
Target
|
tecnología
|
Indicación
|
Efectos biológicos
|
|
Omalizumab
|
Xolair/
Novartis
Pharmaceuticals Corp
|
IgE
|
Hybridoma - Anticuerpo
monoclonal humanizado IgG1 Producido en células ováricas de hámster chino
mediante tecnología de ADN recombinante.
|
-Asma alérgica
persistente moderada o severa en pacientes ≥ 6 años
- Tratamiento de adultos ≥18 años afectados de pólipos nasales
-Urticaria crónica
espontánea en pacientes ≥ 12 años
|
↓ Reducción de la IgE total circulante
Down-regulación de los
receptores FcϵRI en basófilos, mastocitos y células
dendríticas.
|
|
Mepolizumab
|
Nucala /
GlaxoSmithKline
|
IL-5
|
Hybridoma - Anticuerpo
monoclonal humanizado IgG1. Producido en células ováricas de hámster chino
mediante tecnología de ADN recombinante.
|
-Pacientes ≥ 6 años con asma grave eosinofílica
-Pacientes adultos con
granulomatosis
eosinofílica con
poliangeítis
*Aprobado por la FDA
-síndrome hipereosinofílico (HES) durante ≥6 meses sin una causa
secundaria no hematológica identificable ≥ 12 años
-Rinosinusitis crónica con poliposis nasal ≥ 18 años
|
Bloqueo de la unión de
IL-5/IL-5R en eosinófilos
↓ Eosinófilos en sangre
↓ Eosinófilos en esputo
|
|
Benralizumab
|
Fasenra/
AstraZeneca
|
IL-5Rα
|
Hybridoma - Anticuerpo
monoclonal humanizado IgG1.
Producido en células
ováricas de hámster chino mediante tecnología de ADN recombinante.
|
-Pacientes ≥ 12 años con asma eosinofílica grave.
|
↓ Eosinófilos y basófilos a través de la citotoxicidad mediada por células dependiente de anticuerpos
|
|
Dupilumab
|
Dupixent/
Sanofi
|
IL-4Rα
|
Transgenic mice -
IgG4 humana
|
-Pacientes ≥ 6 años con asma grave e inflamación de tipo 2.
-Dermatitis atópica grave en pacientes ≥ 6 meses de edad
-Rinosinusitis crónica con poliposis nasal ≥ 18 años
- Esofagitis eosinofílica en adultos y adolescentes ≥ 12 años
- Pacientes adultos con prurigo nodular ≥ 18 años
|
Bloqueo de la unión de
IL-4/IL-4Rα
Bloqueo de la unión de
IL-13/IL-4Rα
|
|
En fase III de
investigación en Argentina - Aprobados por la FDA (Administración de
Alimentos y Medicamentos)
|
|
Reslizumab
|
Cinqair-Cinqaer/
TEVA
|
IL-5
|
Hybridoma-
Anticuerpo monoclonal
humanizado IgG4. Producido en células de mieloma de ratón
mediante tecnología de
DNA recombinante.
|
Asma con fenotipo eosinofílico grave en pacientes ≥ 18 años
|
Bloqueo de la unión de
IL-5/IL-5R
↓ Eosinófilos circulantes
↓ Eosinófilos en el esputo
|
|
Tezepelumab
|
Tezspire/
AstraZeneca
|
TSLP
|
Hybridoma-Producido en
células de ovario de hámster chino mediante tecnología de ADN recombinante -
IgG2 humana
|
Asma grave en pacientes ≥ 12 años
|
Bloqueo de la unión de
TSLP con su receptor
|
|
Tralokinumab
|
Adtralza, Adbry/
LEO Pharma A/S
|
IL-13
|
Phage display- Anticuerpo monoclonal humano IgG4
|
Dermatitis atópica de moderada a grave ≥ 12 años
|
Bloqueo de la unión de
IL-13 con el receptor IL-13Rα1/IL-4Rα.
|
*Administración
Nacional de Medicamentos, Alimentos y Tecnología Médica de la República
Argentina. Disponible en https://www.argentina.gob.ar/anmat. [última consulta: 17 de noviembre de 2024].
Conclusión
La mejor comprensión de los mecanismos inmunológicos de las
enfermedades alérgicas y el avance en la ingeniería de anticuerpos nos ha
permitido el desarrollo de nuevas terapias biológicas para el tratamiento de
trastornos inflamatorios y alérgicos graves. La indicación de los anticuerpos
monoclonales disponibles debe realizarse bajo rigurosas pautas de selección de
pacientes, con administración constante y con adecuada periodicidad.
En la actualidad disponemos de biológicos mayoritariamente
dirigidos al perfil de endotipo T2. En el futuro necesitamos nuevos desarrollos
de moléculas que tengan como blanco otras vías más allá de las citoquinas TH2
convencionales, así como establecer criterios precisos de remisión de las enfermedades
alérgicas que nos permitan protocolizar, si es posible, cómo y cuándo reducir o
suspender la terapia con biológicos.
1. Winau F, Winau R. Emil von
Behring and serum therapy. Microbes Infect. 2002;4(2):185-8. doi:
10.1016/s1286-4579(01)01526-x.
2. Porter RR. Chemical structure
of gamma-globulin and antibodies. Br Med Bull 1963; 19:197-201. doi:
10.1093/oxfordjournals.bmb.a070056.
3. Köhler G, Milstein C.
Continuous cultures of fused cells secreting antibody of predefined
specificity. Nature. 1975 ;256(5517):495-7. doi: 10.1038/256495a0.
4. Beck A, Wurch T, Bailly C,
Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies.
Nat Rev Immunol. 2010 May;10(5):345-52. doi: 10.1038/nri2747.
5. Todd PA, Brogden RN. Muromonab
CD3. A review of its pharmacology and therapeutic potential. Drugs 1989
;37(6):871-99. doi: 10.2165/00003495-198937060-00004.
6. Khazaeli MB, Conry RM, LoBuglio
AF. Human immune response to monoclonal antibodies. J Immunother Emphasis Tumor
Immunol 1994;15(1):42-52. doi: 10.1097/00002371-199401000-00006
7. Jaffers GJ, Fuller TC, Cosimi
AB, Russell PS, Winn HJ, Colvin RB. Monoclonal antibody therapy. Anti-idiotypic
and non-anti-idiotypic antibodies to OKT3 arising despite intense
immunosuppression. Transplantation. 1986 May;41(5):572-8. doi:
10.1097/00007890-198605000-00004.
8. Abramowicz D, Crusiaux A,
Goldman M. Anaphylactic shock after retreatment with OKT3 monoclonal antibody.
N Engl J Med 1992;327(10):736. doi: 10.1056/NEJM199209033271018.
9. Hwang WY, Foote J.
Immunogenicity of engineered antibodies. Methods. 2005;36(1):3-10. doi:
10.1016/j.ymeth.2005.01.001.
10. Lu RM, Hwang YC, Liu IJ, Lee
CC, Tsai HZ, Li HJ, Wu HC. Development of therapeutic antibodies for the
treatment of diseases. J Biomed Sci. 2020 Jan 2;27(1):1. doi:
10.1186/s12929-019-0592-z.
11. Smith GP. Filamentous fusion
phage: novel expression vectors that display cloned antigens on the virion
surface. Science 1985;228(4705):1315-7. doi:
10.1126/science.4001944.
12. Alfaleh MA, Alsaab HO, Mahmoud
AB, Alkayyal AA, Jones ML, Mahler SM, Hashem AM. Phage display derived
monoclonal antibodies: From bench to bedside. Front Immunol 2020; 11:1986. doi:
10.3389/fimmu.2020.01986.
13. de Marco A. Mechanisms
underlying the efficacy and safety of IgG, antibody fragments, and design
immune biologics. J Allergy Clin Immunol. 2023 Aug;152(2):347-349. doi:
10.1016/j.jaci.2023.01.009.
14. Nur A, Schubert M, Lai JY, Hust
M, Choong YS, Isa WYHW, Lim TS. Antibody Phage Display. Methods Mol Biol.
2023;2702:3-12. doi: 10.1007/978-1-0716-3381-6_1.
15. Lonberg N, Taylor LD, Harding
FA, Trounstine M, Higgins KM, Schramm SR, Kuo CC, Mashayekh R, Wymore K, McCabe
JG, et al. Antigen-specific human antibodies from mice comprising four distinct
genetic modifications. Nature. 1994 Apr 28;368(6474):856-9. doi:
10.1038/368856a0.
16. Green LL, Hardy MC,
Maynard-Currie CE, Tsuda H, Louie DM, Mendez MJ, Abderrahim H, Noguchi M, Smith
DH, Zeng Y, David NE, Sasai H, Garza D, Brenner DG, Hales JF, McGuinness RP,
Capon DJ, Klapholz S, Jakobovits A. Antigen-specific human monoclonal
antibodies from mice engineered with human Ig heavy and light chain YACs. Nat
Genet. 1994 May;7(1):13-21. doi: 10.1038/ng0594-13.
17. Green LL. Antibody engineering
via genetic engineering of the mouse: XenoMouse strains are a vehicle for the
facile generation of therapeutic human monoclonal antibodies. J Immunol
Methods. 1999 Dec 10;231(1-2):11-23. doi: 10.1016/s0022-1759(99)00137-4.
18. Mompó SM, González-Fernández Á.
Antigen-specific human monoclonal antibodies from transgenic mice. Methods Mol
Biol 2019;1904:253-291. doi: 10.1007/978-1-4939-8958-4_11.
19. Jakobovits A, Amado RG, Yang
X, Roskos L, Schwab G. From XenoMouse technology to panitumumab, the first
fully human antibody product from transgenic mice. Nat Biotechnol. 2007
Oct;25(10):1134-43. doi: 10.1038/nbt1337.
20. Pirkalkhoran S, Grabowska WR,
Kashkoli HH, Mirhassani R, Guiliano D, Dolphin C, Khalili H. Bioengineering of
Antibody Fragments: Challenges and Opportunities. Bioengineering (Basel). 2023
Jan 17;10(2):122. doi: 10.3390/bioengineering10020122.
21. Wang Z, Wang G, Lu H, Li H,
Tang M, Tong A. Development of therapeutic antibodies for the treatment of
diseases. Mol Biomed 2022;3(1):35. doi: 10.1186/s43556-022-00100-4.
22. Brekke OH, Sandlie I.
Therapeutic antibodies for human diseases at the dawn of the twenty-first
century. Nat Rev Drug Discov 2003 ;2(1):52-62. doi: 10.1038/nrd984. Erratum in:
Nat Rev Drug Discov. 2003 Mar;2(3):240.
23. Foltz IN, Karow M, Wasserman
SM. Evolution and emergence of therapeutic monoclonal antibodies: what
cardiologists need to know. Circulation 2013;127(22):2222-30. doi:
10.1161/CIRCULATIONAHA.
24. Lobo ED, Hansen RJ, Balthasar
JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci.
2004;93(11):2645-68. doi: 10.1002/jps.20178
25. Lozano NA, Lozano A, Marini V,
Saranz RJ, Blumberg RS, Baker K, Agresta MF, Ponzio MF. Expression of FcRn
receptor in placental tissue and its relationship with IgG levels in term and
preterm newborns. Am J Reprod Immunol. 2018 Sep;80(3):e12972. doi:
10.1111/aji.12972.
26. Wang W, Wang EQ, Balthasar JP.
Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther
2008 ;84(5):548-58. doi: 10.1038/clpt.2008.170.
27. Dostalek M, Gardner I,
Gurbaxani BM, Rose RH, Chetty M. Pharmacokinetics, pharmacodynamics and
physiologically-based pharmacokinetic modelling of monoclonal antibodies. Clin
Pharmacokinet. 2013 Feb;52(2):83-124. doi: 10.1007/s40262-012-0027-4.
28. Castelli MS, McGonigle P,
Hornby PJ. The pharmacology and therapeutic applications of monoclonal
antibodies. Pharmacol Res Perspect 2019 ;7(6):e00535. doi: 10.1002/prp2.535.
29. An G. Concept of Pharmacologic
Target-Mediated Drug Disposition in Large-Molecule and Small-Molecule Compounds.
J Clin Pharmacol. 2020 Feb;60(2):149-163. doi: 10.1002/jcph.1545.
30. Junghans RP. Finally! The
Brambell receptor (FcRB). Mediator of transmission of immunity and protection
from catabolism for IgG. Immunol Res 1997;16(1):29-57. doi: 10.1007/BF02786322.
Erratum in: Immunol Res 1997;16(2):215.
31. Garg A, Balthasar JP.
Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue
kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn 2007;
34(5):687-709. doi: 10.1007/s10928-007-9065-1.
32. Muraro A, Lemanske RF Jr,
Hellings PW, Akdis CA, Bieber T, Casale TB, Jutel M, Ong PY, Poulsen LK,
Schmid-Grendelmeier P, Simon HU, Seys SF, Agache I. Precision medicine in
patients with allergic diseases: Airway diseases and atopic dermatitis-PRACTALL
document of the European Academy of Allergy and Clinical Immunology and the
American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol.
2016 May;137(5):1347-58. doi: 10.1016/j.jaci.2016.03.010.
33. Kuruvilla ME, Lee FE, Lee GB. Understanding asthma
phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol 2019
;56(2):219-233. doi: 10.1007/s12016-018-8712-1.
34.
Durán
González A, Saranz RJ, Lozano NA, Alegre G, Robredo P, Visconti P, Lozano A.
Estado actual del uso de medicamentos biológicos en asma grave. Rev Methodo
2020;5(2):63-69. doi: 10.22529/me.2020.5(2)05
35. Nagase H, Suzukawa M, Oishi K,
Matsunaga K. Biologics for severe asthma: The real-world evidence,
effectiveness of switching, and prediction factors for the efficacy. Allergol
Int. 2023 Jan;72(1):11-23. doi: 10.1016/j.alit.2022.11.008.
36. Hinks TSC, Levine SJ,
Brusselle GG. Treatment options in type-2 low asthma. Eur Respir J
2021;57(1):2000528. doi: 10.1183/13993003.00528-2020.
37. Hynes GM, Hinks TSC. The role
of interleukin-17 in asthma: a protective response? ERJ Open Res 2020
;6(2):00364-2019. doi: 10.1183/23120541.00364-2019.
38. Busse WW, Holgate S, Kerwin E,
Chon Y, Feng J, Lin J, Lin SL. Randomized, double-blind, placebo-controlled
study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in
moderate to severe asthma. Am J Respir Crit Care Med. 2013 Dec
1;188(11):1294-302. doi: 10.1164/rccm.201212-2318OC.
39. Brightling CE, Nair P, Cousins
DJ, Louis R, Singh D. Risankizumab in Severe Asthma - A Phase 2a,
Placebo-Controlled Trial. N Engl J Med. 2021 Oct 28;385(18):1669-1679. doi:
10.1056/NEJMoa2030880.
40. Maurer M, Khan DA, Elieh Ali
Komi D, Kaplan AP. Biologics for the Use in Chronic Spontaneous Urticaria: When
and Which. J Allergy Clin Immunol Pract. 2021 Mar;9(3):1067-1078. doi:
10.1016/j.jaip.2020.11.043.
41. Kaplan A, Lebwohl M, Giménez-Arnau
AM, Hide M, Armstrong AW, Maurer M. Chronic spontaneous urticaria: Focus on
pathophysiology to unlock treatment advances. Allergy. 2023 Feb;78(2):389-401.
doi: 10.1111/all.15603.
42. Muñoz-Bellido FJ, Moreno E, Dávila
I. Dupilumab: A review of present indications and off-label uses. J Investig
Allergol Clin Immunol 2022;32(2):97-115. doi: 10.18176/jiaci.0682.
43. Magerl M, Terhorst D, Metz M,
Altrichter S, Zuberbier T, Maurer M, Bergmann KC. Benefit of mepolizumab
treatment in a patient with chronic spontaneous urticaria. J Dtsch Dermatol
Ges. 2018 Apr;16(4):477-478. doi: 10.1111/ddg.13481
44. Bernstein JA, Singh U, Rao MB,
Berendts K, Zhang X, Mutasim D. Benralizumab for Chronic Spontaneous Urticaria.
N Engl J Med. 2020 Oct 1;383(14):1389-1391. doi: 10.1056/NEJMc2016395.
45. Bartemes KR, Kita H. Roles of
innate lymphoid cells (ILCs) in allergic diseases: The 10-year anniversary for
ILC2s. J Allergy Clin Immunol 2021;147:1531-47. Doi:
10.1016/j.jaci.2021.03.015.
46. Sroka-Tomaszewska J, Trzeciak
M. Molecular mechanisms of atopic dermatitis pathogenesis. Int J Mol Sci
2021;22(8):4130. doi: 10.3390/ijms22084130.
47. Xu Y, Guo L, Li Z, Wu S, Jiang
X. Efficacy and safety profile of dupilumab for the treatment of atopic
dermatitis in children and adolescents: A systematic review and meta-analysis.
Pediatr Dermatol. 2023 Sep-Oct;40(5):841-850. doi: 10.1111/pde.15398
48. Butala S, Paller AS. Biologics
in the management of childhood atopic dermatitis. J Allergy Clin Immunol 2023;
151(3):681-685. doi: 10.1016/j.jaci.2023.01.010.
49. Pelaia C, Pelaia G, Maglio A,
Tinello C, Gallelli L, Lombardo N, Terracciano R, Vatrella A. Pathobiology of
Type 2 Inflammation in Asthma and Nasal Polyposis. J Clin Med. 2023 May
9;12(10):3371. doi: 10.3390/jcm12103371.
50. Mandl HK, Miller JE, Beswick
DM. Current and novel biologic therapies for patients with asthma and nasal
polyps. Otolaryngol Clin North Am. 2023:S0030-6665(23)00153-6. doi:
10.1016/j.otc.2023.08.006.
Conflicto
de interés:
Ninguno.
Limitaciones
de responsabilidad
La
responsabilidad de esta publicación es de los autores.
Fuentes
de apoyo
No
posee.
Originalidad
Este artículo es
original y no ha sido enviado para su publicación a otro medio de difusión
científica en forma completa ni parcialmente.
Cesión de derechos
Quienes participaron en
la elaboración de este artículo, ceden los derechos de autor a la Universidad
Nacional de Córdoba para publicar en la Revista de la Facultad de Ciencias
Médicas de Córdoba y realizar las traducciones necesarias al idioma inglés.
Contribución de los autores
Quienes participaron en
la elaboración de este artículo, han trabajado en la concepción del diseño,
recolección de la información y elaboración del manuscrito, haciéndose
públicamente responsables de su contenido y aprobando su versión final.
Recibido: 2024-02-28 Aceptado: 2024-04-06
 https://creativecommons.org/licenses/by-nc/4.0/
https://creativecommons.org/licenses/by-nc/4.0/
 DOI: http://dx.doi.org/ 10.31053/1853.0605.v81.n4.44413
DOI: http://dx.doi.org/ 10.31053/1853.0605.v81.n4.44413
©Universidad Nacional de Córdoba